Liang’s research explores basic mechanisms underlying fundamental cellular processes in inflammation, infection, and cancer, broadly focusing on autophagy, organelle homeostasis, genomic stability, membrane trafficking, and virus-host interaction.
Liang obtained her M.D. degree from Qingdao University School of Medicine, China, and her Ph.D. degree in genetics from State University of New York (SUNY) at Stony Brook. She received postdoctoral training in tumor virology at Harvard Medical School in the Department of Microbiology and Molecular Genetics. She established her laboratory in 2009 in the Department of Molecular Microbiology and Immunology, University of Southern California, Keck School of Medicine in Los Angeles, where she was promoted to tenured associate professor in 2015. Liang joined The Wistar Institute as a professor in 2020.
The Liang Laboratory

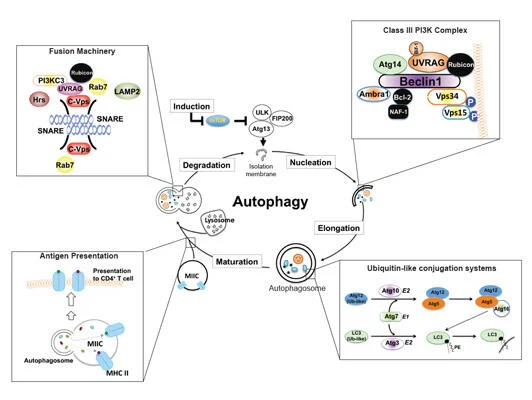
Autophagy (literally “self-eating”) is the natural, regulated mechanism cells use to digest and remove unwanted components. This process influences diverse aspects of cell homeostasis and constitutes a barrier against malignant transformation.
The Liang lab identified a novel autophagy pathway controlled by the tumor suppressor gene UVRAG (UV-radiation Resistance-Associated Gene) that also plays a direct role in DNA repair and chromosomal stability. The group studies autophagy and related pathways in leukemia, colorectal cancer, melanoma pathogenesis and therapy resistance, and in viral persistency.

-
Postdoctoral Fellows
Jinghui Liang, Ph.D.
Behzad Mansoori, Ph.D.
Christian Pangilinan, Ph.D
Qing Zhu, Ph.D. -
Research Assistants
Nivedita Rathaur
Solmaz Shirjang -
Bioinformatics Technicians
Robert McElroy
Jonathan Richards -
Predoctoral Trainees
Dongliang Shen
Lu Zhang -
Lab Manager
Dali Nemecio
-
Liang Jin, Ph.D. (2010) Senior Scientist, China
Zhen Zhao, Ph.D. (2010 – 2012) Assistant Professor, University of Southern California
Lingling Shi, Ph.D. (2011 – 2012) Associate Professor, Guangdong-Hong Kong-Macau Institute of CNS Regeneration, China
Shanshan He, M.D., Ph.D. (2012 – 2015) Senior Scientist, NanoString Technologies, Inc.
Yongfei Yang (2013 – 2017) Associate Professor, Beijing Institute of Technology
Gym-beom Jang (2016 – 2018) Staff Researcher, National Cancer Center, Seoul, Korea
Shun Li, Ph.D. (2016 – 2018) Professor, Chengdu Medical School, Sichuan University
Hongrui Guo (2016 – 2018) Professor, Sichuan Agriculture University
Ying Song (2017-2020) Professor, Weifang Medical College
-
Motivated recent Ph.D. graduates from related fields with a record of publications are encouraged to inquire about the position below. Contact cliang@wistar.org.
– Postdoctoral Fellow
Research

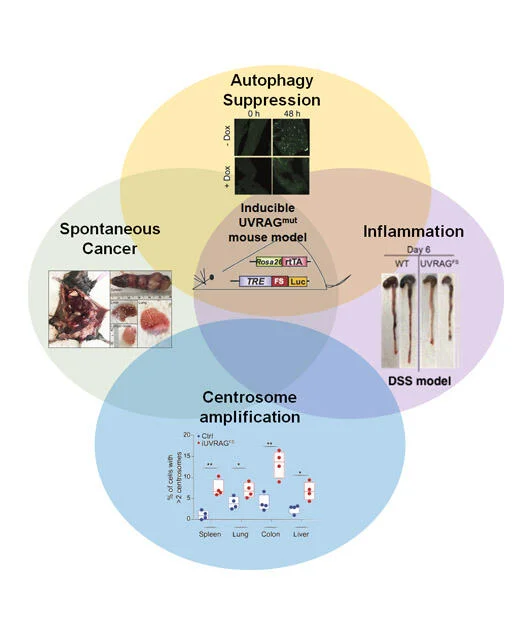
Macroautophagy/autophagy deregulation has been observed in perpetuated inflammation and the proliferation of tumor cells. However, the mechanisms underlying these changes have yet to be well-identified. UVRAG is one of the key players of autophagy, but its role in vivo remained puzzling. Our recent study, published in Nature Communications (Quach, Song et al), utilized a mouse model with inducible expression of a cancer-derived frameshift (FS) mutation in UVRAG that dominant-negatively inhibits wild-type UVRAG, resulting in impaired stimulus-induced autophagy. The systemically compromised autophagy, particularly mitophagy, notably increases inflammation and associated pathologies. Another pivotal result of this study is that it uncovered a “missing molecular link” between age-related decreases in autophagy and aging-induced spontaneous cancers, which could provide new insight into how basal autophagy protects against cancer. Using our mouse model, we observed that suppression of UVRAG accelerated age-related decline in autophagy and impaired autophagic turnover of oncogenic β-catenin, leading to increased proliferation and tumor formation in mice. It remains to be seen whether this age-dependent regulation of β-catenin by autophagy will translate to other autophagy-related cancer models. Nevertheless, our work represents a key advance in understanding how UVRAG and by extension autophagy works in vivo.
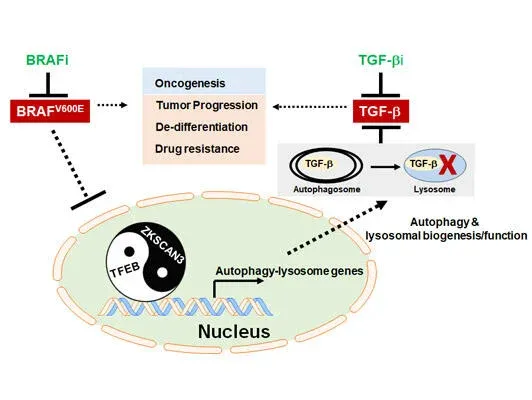
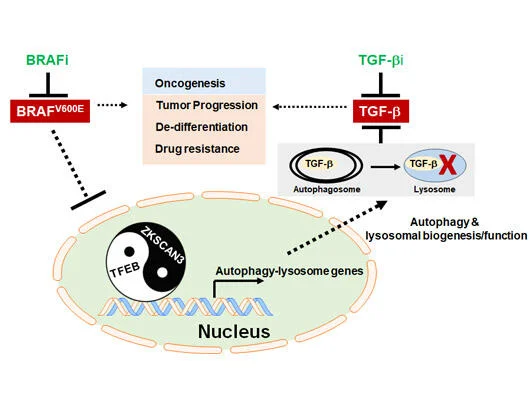
Autophagy, originally described as a lysosome-dependent degradation of cytoplasmic components upon starvation, has since been shown to influence diverse aspects of homeostasis, constituting a barrier against malignant transformation. Despite its inhibitory role in tumor initiation, autophagy is postulated to fuel the growth of established tumors and confers drug resistance, principally as a survival mechanism. In melanoma, where 40–60% of cases have a mutation in BRAF (BRAFV600E), conflicting results have been reported regarding the relationship between autophagy and the mutant, and the interaction between BRAF signaling and autophagy has been ambiguous. Our work, published in 2019 in Nature Communications (Li, et al), demonstrated that in BRAFV600E-melanoma autophagy is induced by BRAF inhibitor (BRAFi), as part of a transcriptional program coordinating lysosome biogenesis/function, mediated by the TFEB transcription factor. TFEB is phosphorylated and thus inactivated by BRAFV600E via its downstream ERK independently of mTORC1. BRAFi disrupt TFEB phosphorylation, allowing its nuclear translocation, which is synergized by increased phosphorylation/inactivation of the ZKSCAN3 transcriptional repressor by JNK2/p38-MAPK. Blockade of BRAFi-induced transcriptional activation of autophagy-lysosomal function in melanoma xenografts causes enhanced tumor progression, EMT-transdifferentiation, metastatic dissemination, and chemoresistance, which is associated with elevated TGF-β levels and enhanced TGF-β signaling. Inhibition of TGF-β signaling restores tumor differentiation and drug responsiveness in melanoma cells. Thus, the “BRAF-TFEB-autophagy-lysosome” axis represents an intrinsic regulatory pathway in BRAF-mutant melanoma, coupling BRAF signaling with TGF-β signaling to drive tumor progression and chemoresistance.
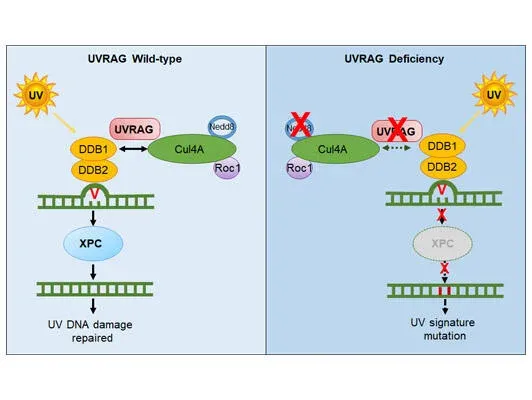
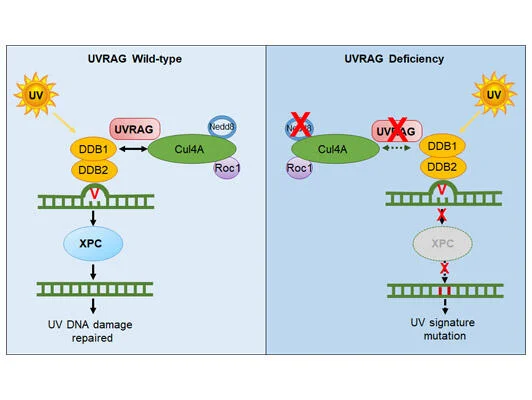
Malignant melanoma ranks as the number one cause of death from skin cancers. One striking feature of cutaneous melanoma is the highest mutation burden caused by ultraviolet radiation (UVR) in its genome. Such high UV-signature mutation rate provides huge fitness advantage to cancer cells. However, the molecular mechanisms underlying UV-induced mutagenesis in melanoma remain largely unknown, posing major challenges in the development of cancer treatment with long-term durability of responses. While screening for the molecular target(s) that drive UV signature loads in melanoma genomes, we discovered that UV-exposed skin melanoma patients with higher amounts of UV signature in their genomes had reduced levels of UVRAG; but this was not observed for UV-shielded melanoma, suggesting a potential link of UVRAG to photolesion protection. Our work, published in 2016 in Molecular Cell (Yang et al), further demonstrated that UVRAG plays an integral role in UV-induced DNA damage repair. It localizes to photolesions and associates with DDB1 to promote the assembly and activity of the DDB2-DDB1-Cul4A-Roc1 (CRL4DDB2) ubiquitin ligase complex, leading to efficient XPC recruitment and global genomic NER. UVRAG depletion decreased substrate handover to XPC and conferred UV-damage hypersensitivity. These results identify UVRAG as a regulator of CRL4DDB2-mediated NER and suggest that its expression levels may influence melanoma predisposition. Future studies in the laboratory will focus on investigating the mechanisms protecting against UV-induced mutagenesis, and delineate why they fail to work in melanoma, enabling early risk prediction and prognostication, inspiring new strategy to minimize melanoma susceptibility and tumor fitness.
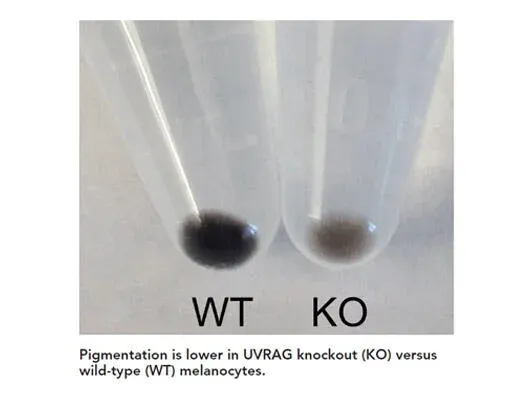
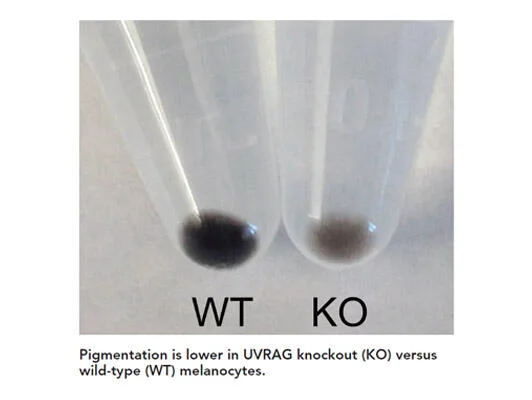
Skin pigmentation provides first-line protection against UV radiation (UVR) that increases the risk of skin cancers. However, mechanisms underlying this process remain poorly understood. Our work, published in 2018 in PNAS (Yang, Jang et al), identified the autophagic tumor suppressor UVRAG as a bona fide player in melanosome biogenesis by targeting biogenesis of lysosome-related organelles complex 1 (BLOC-1) independently of autophagy. UVRAG maintains the localization and stability of BLOC-1 to facilitate the sorting/delivery of melanogenic cargoes. Reduced levels of UVRAG rendered cells unresponsive to UVR–α-MSH–MITF signaling and defective melanocyte development in vivo. Moreover, UVRAG-mediated melanogenesis and tanning response were impaired in oncogene driven melanoma. This study represents a description of a noncanonical role of autophagy factor in melanogenic remodeling and also provides mechanistic insights into UVRAG in pigmentation disorder and UV-associated cancer. Future studies will continue to investigate the molecular mechanisms controlling melanosome biogenesis/dynamics and their impact on UV protection in skin cancer.
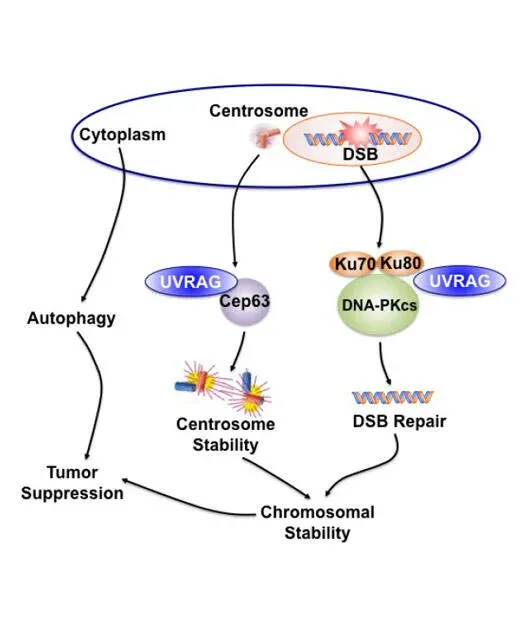
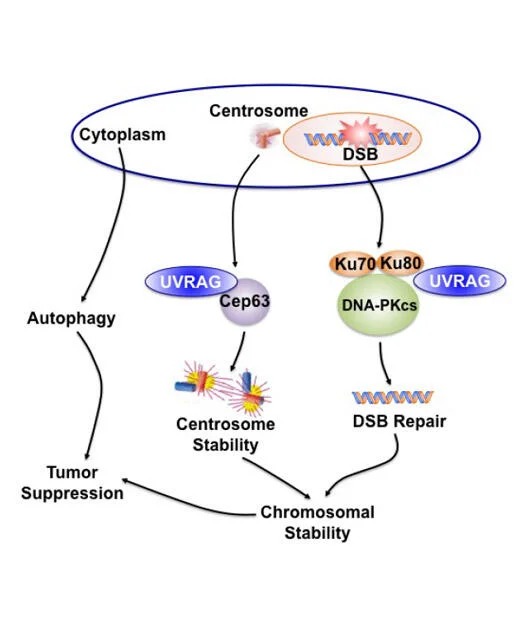
Due to the unacceptable toxicity and ineffectiveness of currently cancer chemotherapy and radiation, the search for new molecular targets is in high gear. Chromosomal instability (CIN) is a hallmark of most aggressive cancers. However, efforts to exploit CIN therapeutically have been hampered by a lack of understanding of molecular mechanisms involved in its regulation. Our objective is to characterize the molecular mechanisms that induce CIN and to identify new therapeutic targets for cancer progression by applying our discovery of a novel molecule in CIN regulation. While we initially thought that UVRAG was mainly a promoter of the autophagy pathway, our continued efforts demonstrated that, in addition to its confirmed role in autophagy activation, UVRAG also plays a direct role in preventing cells from accumulating abnormal chromosomes, which would increase the danger of developing oncogenic mutations. As the maintenance of chromosomal integrity is a fundamental biological process to thwart tumor formation, this property of UVRAG also explains why it is frequently mutated in cancers. Our work, published in 2012 in Developmental Cell (Zhao, Oh et al) have discovered two novel mechanisms by which UVRAG mediates nonhomologous end-joining (NHEJ) and centrosome stability in a spatiotemporally distinct manner, independently of autophagy. These findings will help us expand on this knowledge to investigate in depth how UVRAG impacts genomic integrity and cancer development, and determine the molecular crosstalk between the autophagy machinery and the genome surveillance process of the cells.
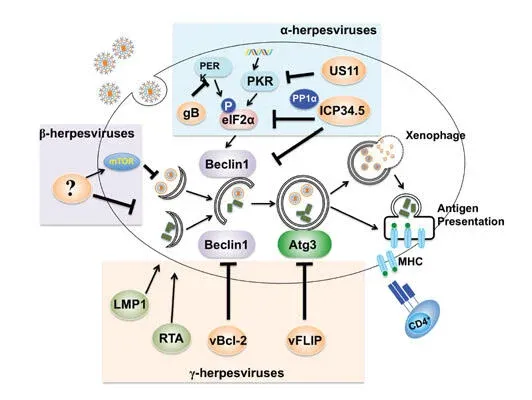
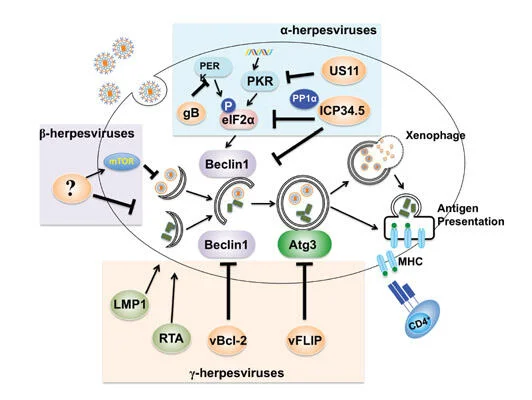
Bcl-2, originally identified as a universal inhibitor of apoptotic cell death, has since been implicated in suppressing autophagy, the cell’s quality control mechanism. Our study, published in 2011 in Cell Death and Differentiation (Oh et al), demonstrates that the anti-autophagic aspect of Bcl-2 can function as a promoter of oncogenic growth, independently of its role in apoptosis signaling. It is likely that the increase in Bcl-2 often seen in breast and other cancers might render cells error prone by blunting autophagy, while concomitantly keeping damaged cells alive. The outcome of such a ‘double hit’ of Bcl-2 may synergistically promote tumor growth and increase the chance of cancer development and drug resistance. Moreover, our lab has identified the mechanism by which viruses co-opt the autophagic pathway to establish virulence in vivo. Published in 2009 in PLOS Pathogens (E et al), we presented direct in vivo evidence for the key role of the anti-autophagic aspect of the virally encoded Bcl-2s in the chronic infection of oncogenic gamma-herpesviruses, and proposed that cellular autophagy may have a substantial effect on viral persistence and may influence the in vivo fitness of viruses. This discovery expands upon known antiviral activities of the autophagy machinery, and also suggests new approaches for treating some virally induced diseases.
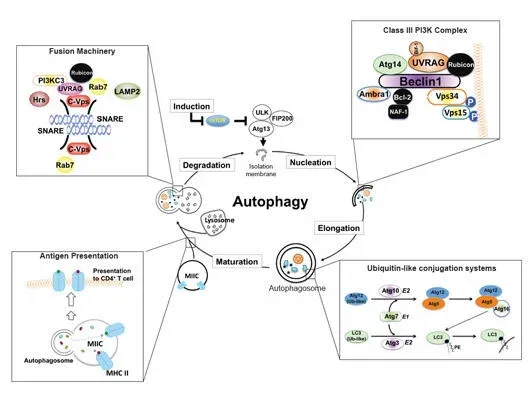
The homeostasis of all eukaryotic cells depends on their “greening” ability to use a lysosomal pathway known as autophagy to degrade and recycle self-components. Our work, published in 2008 and 2013 in Nature Cell Biology (He, et al), have shown for the first time that autophagic and endosomal maturation are of topological similarity and may have evolved to share associated machinery for lysosomal delivery, which has been further confirmed by other laboratories later on. Our seminal findings showed that UVRAG, originally identified as a UV-protecting molecule, is an important coordinator in autophagic and endosomal trafficking through its interaction with the class C Vps tethering complex. Furthermore, UVRAG recognizes PI3P at the ER, where it couples the ER tether to govern Golgi-ER retrograde transport. Intriguingly, when autophagy is induced, UVRAG undergoes a “partnering shift” from the ER complex to the BECN1 autophagy complex, resulting in concomitant inhibition of Golgi-ER transport and activation of ATG9 autophagic trafficking. This work sets up a mechanism of how cells achieve spatiotemporal fidelity of protein transport and organelle homeostasis, providing insights into trafficking-related diseases.
-
Postdoctoral Fellows
Jinghui Liang, Ph.D.
Behzad Mansoori, Ph.D.
Christian Pangilinan, Ph.D
Qing Zhu, Ph.D. -
Research Assistants
Nivedita Rathaur
Solmaz Shirjang -
Bioinformatics Technicians
Robert McElroy
Jonathan Richards -
Predoctoral Trainees
Dongliang Shen
Lu Zhang -
Lab Manager
Dali Nemecio
-
Liang Jin, Ph.D. (2010) Senior Scientist, China
Zhen Zhao, Ph.D. (2010 – 2012) Assistant Professor, University of Southern California
Lingling Shi, Ph.D. (2011 – 2012) Associate Professor, Guangdong-Hong Kong-Macau Institute of CNS Regeneration, China
Shanshan He, M.D., Ph.D. (2012 – 2015) Senior Scientist, NanoString Technologies, Inc.
Yongfei Yang (2013 – 2017) Associate Professor, Beijing Institute of Technology
Gym-beom Jang (2016 – 2018) Staff Researcher, National Cancer Center, Seoul, Korea
Shun Li, Ph.D. (2016 – 2018) Professor, Chengdu Medical School, Sichuan University
Hongrui Guo (2016 – 2018) Professor, Sichuan Agriculture University
Ying Song (2017-2020) Professor, Weifang Medical College
-
Motivated recent Ph.D. graduates from related fields with a record of publications are encouraged to inquire about the position below. Contact cliang@wistar.org.
– Postdoctoral Fellow



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}